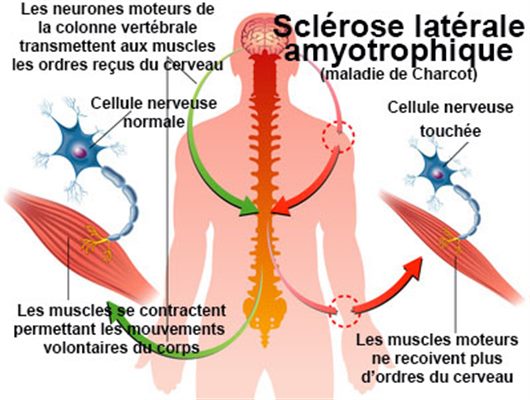
La maladie de Charcot ou sclérose latérale amyotrophique (SLA) est une maladie neurodégénérative rare se traduisant par une atrophie musculaire progressive. De pronostic très sombre, la SLA a été décrite pour la première fois dans les années 1860 par le neurologue français Jean‑Martin Charcot.
150 ans plus tard, son origine demeure mal comprise, et aucun traitement n’a encore été découvert. Les recherches en neurologie ont cependant fait progresser les connaissances sur cette pathologie, et plusieurs pistes thérapeutiques sont en cours d’étude.
Une maladie dégénérative aux origines mal comprises
On estime que la SLA affecte actuellement 7 000 à 8 000 personnes en France. Chaque année, 1 000 nouveaux cas sont diagnostiqués dans notre pays, ce qui représente près de quatre nouveaux patients par jour.
Cette maladie, qui débute aux alentours de 55 ans en moyenne, affecte principalement les neurones moteurs, qui sont responsables du mouvement, tant au niveau « central » (neurones moteurs partant du cerveau et cheminant dans la moelle épinière) qu’au niveau « périphérique » (neurones moteurs partant de la moelle pour former les nerfs et activer les muscles). Chez les patients atteints, ces neurones dégénèrent, ce qui se traduit par une diminution progressive de la force au niveau des bras et des jambes, ainsi qu’au niveau des muscles permettant de parler, d’avaler ou bien de respirer.
Le pronostic de la maladie est malheureusement toujours très sombre, la durée médiane de survie après les premiers signes cliniques variant de 3 à 5 ans.
Neuf fois sur dix, la SLA est « sporadique », c’est-à-dire qu’elle survient sans qu’un autre cas n’ait été détecté dans l’entourage familial. L’origine est alors inconnue. Représentant 10 % des cas, les formes « familiales » de la maladie sont quant à elles liées à une mutation dans un gène déterminé. Plus de 30 gènes ont actuellement été incriminés dans ces formes héréditaires de SLA, quatre d’entre eux (C9ORF72, SOD1, TARBP, FUS) rendant compte de plus de 50 % de ces cas.
Un manque de médicaments
La physiopathologie de la maladie, son origine et ses mécanismes à l’échelle moléculaire et cellulaire restent encore aujourd’hui mal compris. En outre, la grande hétérogénéité clinique observée – la maladie évolue de façon très variable d’un patient à l’autre – est également difficile à expliquer.
L’un des problèmes auxquels se heurtent les scientifiques est que les modèles d’étude actuels de la SLA (cultures de cellules ou souris transgéniques) ne reproduisent qu’imparfaitement la maladie, ce qui complique l’analyse de ses mécanismes et l’évaluation de nouvelles voies thérapeutiques.
La mise au point de nouveaux traitements est donc particulièrement difficile, ce qui explique qu’à l’heure actuelle, seul un médicament – le Riluzole – soit disponible. Malheureusement, si cette molécule a montré son efficacité pour ralentir l’évolution de la maladie, elle ne stoppe pas sa progression.
Des avancées dans le champ des neurosciences opérées depuis 20 ans ont cependant permis d’accumuler de nouvelles connaissances sur la SLA. On pense aujourd’hui que la neurodégénérescence des motoneurones est probablement multifactorielle, mêlant des facteurs génétiques et environnementaux, une neuro-inflammation, des troubles métaboliques, ainsi qu’une toxicité liée à la présence d’agrégats protéiques endogènes. En effet, comme dans d’autres maladies neurodégénératives telles que les maladies d’Alzheimer ou de Parkinson, dans la quasi-totalité des cas de SLA, on constate une agrégation de protéines dans les cellules.
Dans le cas de la SLA, la protéine concernée ̑s’appelle TDP-43. Principalement présente dans le noyau, elle joue un rôle important dans la régulation de l’ARN, une molécule dont la structure est proche de celle de l’ADN et qui joue divers rôles majeurs dans la cellule. Dans certaines conditions, TDP-43 est modifiée et sort du noyau cellulaire pour aller s’accumuler dans le cytoplasme (la région comprise entre la membrane de la cellule et le noyau). On pense que cette accumulation pourrait perturber le bon fonctionnement de la cellule et être responsable de la dégénérescence des neurones. La propagation de la forme anormale de la protéine se fait de proche en proche.
À partir de ces constatations, plusieurs essais thérapeutiques se sont construits ces dernières années, tentant d’agir sur l’une ou l’autre de ces composantes.
Les pistes à l’étude
Parmi ces essais cliniques, l’étude Mirocals propose d’utiliser de faibles doses l’interleukine 2 pour moduler les voies de la neuro-inflammation. L’interleukine 2 est une molécule présente de façon naturelle dans l’organisme, qui augmente le nombre de cellules immunitaires appelées cellules « T régulatrices ». Or, celles-ci ont une action protectrice à l’encontre des réponses immunitaires nocives pour le neurone.
D’autres travaux ont adopté une approche plus métabolique. C’est notamment le cas de ceux portant sur le Masitinib, une molécule capable de réguler la prolifération des astrocytes (des cellules qui apportent aux neurones nutriments et oxygène), et qui a de ce fait un effet neuroprotecteur. Une première étude (AB 10015) réalisée chez 394 patients sur 48 semaines a montré que le Masitinib associé au Riluzole permettait de ralentir l’évolution de la maladie et une étude internationale de phase 3 vient de débuter pour confirmer ces résultats.
Les progrès remarquables et rapides sur la découverte des gènes associés à la SLA, ont également ouvert la porte à l’ère de la thérapie génique pour traiter les formes génétiques de SLA. Actuellement, les traitements à l’étude ont pour objectif de limiter la toxicité des protéines mutées responsables de ces formes de la maladie en inhibant l’expression de leur gène.
Dans cette optique, des essais cliniques sont en cours pour évaluer la sécurité et l’efficacité du Tofersen, un oligonucléotide capable de se fixer spécifiquement sur l’ARN messager d’une de ces protéines afin d’en inhiber la traduction et de réduire sa production (ndlr : dans le noyau de la cellule, l’information correspondant à une protéine donnée, portée par l’ADN, est « recopiée » en ARNm, lequel passe ensuite dans le cytoplasme où il va servir de « plans de montage » pour fabriquer ladite protéine).
Une première étude a été publiée en 2020 dans la revue New England Journal of Medicine, montrant une tendance au ralentissement du déficit moteur et de l’atteinte respiratoire chez les patients ayant reçu 100 mg de Tofersen durant trois mois. Ces résultats encourageants doivent être confirmés lors d’une étude de phase 3 actuellement en cours.
Enfin, ces dernières années plusieurs équipes se sont intéressées au rôle possible d’un déséquilibre du microbiote intestinal dans l’apparition et la progression des maladies neurodégénératives, dont la SLA. Des résultats obtenus sur des souris modèles pour la SLA, publiés en 2019 dans la revue Nature, ont révélé en évidence une altération du microbiote intestinal avant même l’apparition des signes cliniques de la maladie. Ces travaux ont fait naître l’hypothèse d’une potentielle interaction intestin-cerveau pouvant être impliquée dans le développement de la SLA. Une étude publiée en 2021 a aussi mis en évidence des différences entre les microbiotes des patients SLA par rapport à celui de leur conjoint. Ces données récentes suggèrent que la modification de la composition des microbiotes intestinaux pourrait être une piste thérapeutique à explorer.
Ces résultats, ainsi que les avancées des recherches fondamentales et cliniques basées sur les thérapies cellulaires et géniques, ouvrent de nouvelles perspectives : la décennie qui débute pourrait voir l’essor de la médecine personnalisée pour la prise en charge de la SLA.
Source: www.theconversation.com
No responses yet